
Validation Of A RIG-I–Based Functional Assay For Quantitative Detection Of Double-Stranded RNA Impurities In mRNA Drug Substance
By Mohamad Toutounji, Ph.D., founder and principal scientist, Molgenium GmbH

Double-stranded RNA (dsRNA) is a recognized process-related impurity in mRNA drug substances produced by in vitro transcription (IVT) with T7 RNA polymerase. Established release methods, principally J2 antibody dot-blot and dsRNA-specific ELISA, quantify dsRNA mass but do not directly report on its capacity to activate cytosolic pattern recognition receptors. This article describes a functional assay that uses recombinant human RIG-I as a biochemical sensor for dsRNA.
dsRNA As A Process-Related Impurity In IVT mRNA
dsRNA is generated as a side product during IVT of single-stranded mRNA by bacteriophage T7 RNA polymerase. Three mechanisms have been documented in the literature:
- Turn-around (cis) transcription: The 3′ end of the run-off RNA folds back on itself and is extended in cis through the RNA-dependent RNA polymerase activity of T7, producing hairpin dsRNA.10
- Random priming of abortive transcripts: Short abortive transcripts (typically below 10 nt) anneal back onto run-off RNA, in cis or in trans, and are extended into double-stranded products.
- Promoter-independent antisense transcription: T7 RNA polymerase initiates non-canonically from the DNA template terminus, producing antisense RNA that anneals with the sense product to form full-length dsRNA.
Empirical data indicate that even highly optimized IVT processes generate measurable dsRNA, and that dsRNA can persist into purified drug substance unless removed by post-IVT purification steps such as reverse-phase HPLC or cellulose chromatography.1,5
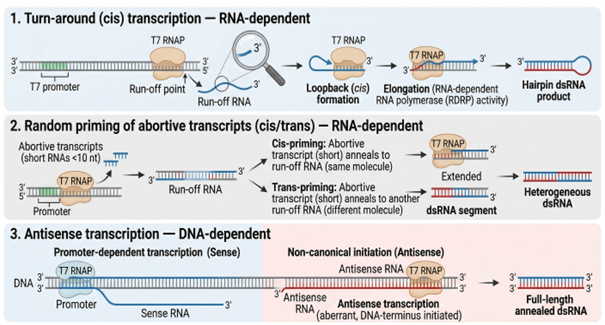
Figure 1. Schematic representation of the three documented mechanisms by which T7 RNA polymerase generates dsRNA during in vitro transcription: (1) cis-acting turn-around transcription producing hairpin dsRNA, (2) random priming of abortive transcripts onto run-off RNA in cis or trans yielding heterogeneous duplexes, and (3) DNA-dependent antisense transcription initiated from the template terminus producing full-length annealed dsRNA. Adapted from author's presentation, AMM/USP Workshop, April 2026.
Biological Consequences And CQA Classification
The biological relevance of residual dsRNA in IVT mRNA drug substance derives from two parallel signaling pathways within human cells.
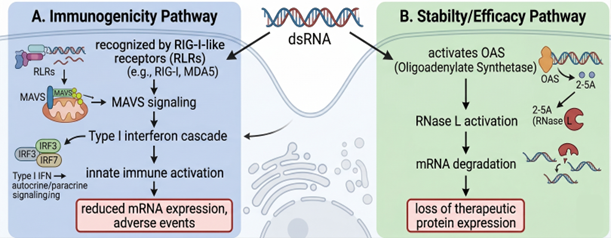
Figure 2. Schematic of the two parallel pathways activated by cytosolic dsRNA. Pathway A (immunogenicity): recognition by RIG-I-like receptors (RIG-I, MDA5) leads to MAVS aggregation at the mitochondrial outer membrane, activation of the TBK1/IKK axis, and induction of type I interferons via IRF3/IRF7. Pathway B (stability/efficacy): activation of 2′,5′-oligoadenylate synthetase (OAS) generates 2-5A, which activates latent RNase L, leading to cleavage of cellular RNA including the therapeutic mRNA. The two pathways converge on reduced therapeutic protein expression and the potential for adverse innate immune events. Adapted from author's presentation, AMM/USP Workshop, April 2026.
The first pathway is innate immune activation through the RIG-I-like receptor (RLR) family. RIG-I (DDX58) and MDA5 (IFIH1) recognize cytosolic dsRNA with structural features characteristic of viral replication intermediates. Blunt 5′ ends bearing a 5′-triphosphate group are particularly potent ligands for RIG-I, while longer duplexes are preferentially recognized by MDA5.2,7,8 Recognition is followed by recruitment of MAVS at the mitochondrial outer membrane, activation of TBK1 and IKK, and induction of type I interferons and pro-inflammatory cytokines via IRF3, IRF7, and NF-κB.
The second pathway is direct interference with translation through OAS/RNase L. Cytosolic dsRNA activates 2′,5′-oligoadenylate synthetase (OAS), generating 2′,5′-oligoadenylates that activate latent RNase L. Activated RNase L cleaves cellular RNA, including the therapeutic mRNA itself, attenuating expression of the encoded protein.
For an IVT-derived mRNA drug substance, residual dsRNA therefore represents a quality attribute with the potential to affect both safety (innate immune activation, reactogenicity) and efficacy (suppression of therapeutic protein expression). Reduction of dsRNA content has been shown to reduce the immunogenicity of nucleoside-modified mRNA and to increase translational output.4,5 On the basis of these mechanistic and experimental data, dsRNA is treated as a critical quality attribute (CQA) within ICH Q8/Q9/Q11 quality by design frameworks for IVT mRNA, requiring quantitative control.
Established Detection Methods And Their Analytical Scope
Two methods are in routine use for dsRNA detection in IVT mRNA matrices.
The J2 monoclonal antibody, originally raised against dsRNA structures, is used in dot-blot and immunohistochemistry formats.9 The method is qualitative or, at best, semi-quantitative; sensitivity depends on duplex length and is reduced for short dsRNA species (<60 bp) and binding is reduced for duplexes containing certain modified nucleosides, including N1-methyl-pseudouridine.
Sandwich ELISA formats using anti-dsRNA antibodies provide quantitative readouts in 96-well plate format with improved tolerance for modified nucleotides relative to dot-blot and are compatible with GMP environments. ELISA does not, however, report on the immunological competence of the detected dsRNA: detection depends on antibody-epitope recognition, not on the capacity of the duplex to engage cellular pattern recognition receptors. Two samples with comparable ELISA signal can therefore differ in their capacity to activate RIG-I or MDA5 in vitro and to induce a type I interferon response in vivo.
Both methods are useful within their respective scopes. They do not directly address the biological readout that drives the in vivo consequences described in the previous section.
Rationale For RIG-I–Based Functional Readout
RIG-I is the cytosolic pattern recognition receptor that initiates the human innate immune response to cytosolic dsRNA. In its resting state, RIG-I is autoinhibited; the helicase 2i (Hel2i) gate sequesters the tandem CARD signaling domains. Binding to a dsRNA ligand bearing a blunt 5′ end and a 5′-triphosphate (or 5′-diphosphate) triggers an ATP-dependent conformational rearrangement, release of the CARDs, and oligomerization on the dsRNA strand.6 Selectivity for genuine dsRNA over host single-stranded RNA exceeds three orders of magnitude. Activation efficiency is also length-dependent: short dsRNA (<500 bp) supports productive RIG-I signaling, while longer duplexes are preferentially recognized by MDA5.
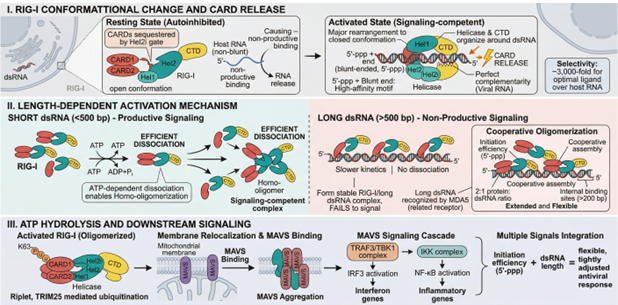
Figure 3. Schematic of RIG-I activation. (I) In the resting state, the tandem CARD domains are sequestered by the Hel2i gate; binding to a blunt-ended, 5′-triphosphorylated dsRNA ligand drives a major conformational rearrangement and CARD release. (II) Length-dependent activation: short dsRNA supports productive signaling through ATP-dependent dissociation and homo-oligomerization, while long dsRNA is preferentially recognized by MDA5. (III) ATP hydrolysis is coupled to membrane relocalization, MAVS aggregation, and downstream IRF3/NF-κB activation. The ATP→ADP conversion is the property exploited by the assay described here. Adapted from author's presentation, AMM/USP Workshop, April 2026.
The ATPase activity of RIG-I is mechanistically coupled to productive dsRNA binding. ATP hydrolysis is not stimulated by ssRNA, ssDNA, or dsDNA at concentrations relevant to mRNA drug substance matrices. ADP production is therefore stoichiometrically linked to dsRNA recognition and provides the basis for a quantitative biochemical readout.

Assay Description
The assay is performed in 96-well plate format. Samples are incubated with recombinant human RIG-I and ATP at 37 degrees C for 60 minutes, during which productive dsRNA binding drives hydrolysis of ATP to ADP. A first reagent depletes residual ATP. A second reagent, comprising pyruvate kinase and phosphoenolpyruvate, regenerates ATP from ADP stoichiometrically. ATP is detected by an optimized luciferase/luciferin reaction, and luminescence is measured on a standard plate luminometer. Sample dsRNA concentration is back-calculated from a reference standard curve, fitted with a five-parameter logistic (5-PL) model consistent with the saturable kinetics of the underlying enzyme reaction.
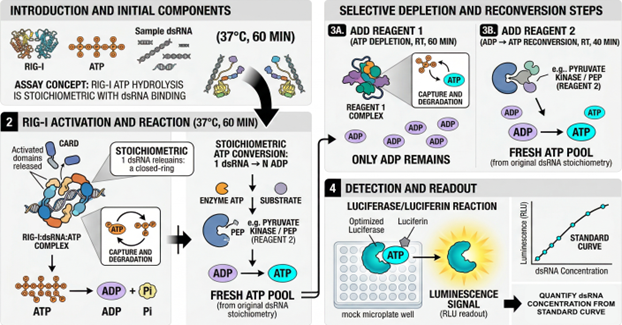
Figure 4. Schematic workflow. (1) Sample is incubated with recombinant RIG-I and ATP; productive dsRNA binding drives ATP hydrolysis to ADP. (2) Reagent 1 depletes residual ATP, leaving ADP proportional to dsRNA-driven activity. (3) Reagent 2 (pyruvate kinase/phosphoenolpyruvate) regenerates ATP stoichiometrically from ADP. (4) Regenerated ATP is detected by a luciferase/luciferin reaction and luminescence is read on a standard plate luminometer. dsRNA concentration is back-calculated from a 5-PL reference curve. Adapted from author's presentation, AMM/USP Workshop, April 2026.
Method Validation: ICH Q2(R2)-Aligned Results
Validation followed an analytical target profile defined a priori. The analyte was specified as dsRNA ≥ 30 bp; matrices included IVT reaction buffer, purified mRNA drug substance, and multiple formulation buffers; nucleotide chemistries covered UTP, pseudouridine triphosphate (pUTP), N1-methyl-pseudouridine triphosphate (N1-Me-pUTP), and 5-methoxyuridine triphosphate (5-OMe-UTP). Acceptance criteria were aligned with ICH Q2(R2) for quantitative impurity procedures (intra-assay CV ≤ 15%, inter-assay CV ≤ 20%, spike recovery 80%–120%).
Critical Reagent Qualification
Recombinant human RIG-I was purified by sequential Ni-NTA affinity chromatography (50 to 75 mM imidazole gradient) and Q-FF anion exchange chromatography (200 to 300 mM NaCl gradient). Identity and purity were confirmed by SDS-PAGE and anti-His Western blot, showing a single dominant band at the expected molecular mass of approximately 106 kDa. dsRNA-dependent ATPase activity was verified for each lot. The two-step purification mitigates co-purification of nucleic acids that could otherwise contribute background ATPase signal.
Specificity
Cross-reactivity was evaluated against ssRNA, ssDNA, and dsDNA at concentrations representative of the mRNA drug substance matrix. At 2.5 ng/µL, dsRNA produced approximately 300,000 RLU, while ssRNA, ssDNA, and dsDNA each produced approximately 40,000 RLU at or near assay background. Back-calculated values for the non-target species fell below 0.02 ng/µL, indicating that the matrix components evaluated do not generate quantifiable false-positive signals under the conditions tested.
Range, Response, And Model Fit
Standard curves were generated with a 300 bp N1-Me-pUTP dsRNA reference standard from 0.039 to 2.5 ng/µL. The response was nonlinear and saturable, consistent with the kinetics of RIG-I ATPase activity. A 5-PL fit produced r² = 0.9997 across the working range. The nonlinear response is consistent with ICH Q2(R2) provisions for procedures based on enzyme kinetics or biological response.
Accuracy
Spike-recovery experiments at three concentrations (0.039, 0.312, and 1.25 ng/µL, each in triplicate) yielded recoveries between 80% and 108%, within the ICH Q2 envelope of 80%–120% at all levels.
Repeatability (Intra-Assay Precision)
Coefficients of variation across the working range were 1%–10%, below the 15% acceptance threshold for impurity methods. Recoveries calculated from triplicate measurements ranged from 93% to 104%.
Intermediate Precision (Inter-Assay)
Intermediate precision was assessed using three independent luciferase reagent batches, identified as the most variable critical reagent in the workflow. Coefficients of variation across the three batches ranged from 4.1% to 14.2%.
Robustness
Method transfer was demonstrated between two laboratories using different luminometers (Shanpu and SpectraMax iD5). Absolute luminescence values differed, as expected from instrument-specific photon detection characteristics, while standard curve shape and back-calculated quantitative results were preserved across the range.
Discussion
The data presented support the use of a RIG-I–based ATPase readout as a quantitative, ICH Q2(R2)-aligned method for dsRNA detection in IVT-derived mRNA drug substance. The method captures a defined biochemical event (productive engagement of a pattern recognition receptor) that mass-based detection does not directly report. The two methodological approaches are therefore orthogonal: ELISA reports total dsRNA epitope mass, while the RIG-I assay reports dsRNA capable of activating an innate immune sensor that is structurally and functionally homologous to the receptor expressed in human cells.
The method has defined boundaries. It has been validated for IVT-produced linear mRNA in the approximate length range 500 to 5,000 nucleotides, with the four nucleotide chemistries listed above (UTP, pUTP, N1-Me-pUTP, 5-OMe-UTP). It has not been evaluated for self-amplifying RNA (saRNA), circular RNA, dsRNA therapeutics, or constructs incorporating backbone or base chemistries outside this set.
Concordance with in vivo immunogenicity readouts is supported by the underlying biology of RIG-I but has not been established quantitatively in head-to-head comparison studies within this validation campaign. Such studies, including comparison with cell-based interferon reporter assays and where appropriate with non-clinical reactogenicity data, represent a relevant area for further work.
Two ICH Q2(R2) parameters remain to be formally completed: the limit of detection (LOD) and the limit of quantitation (LOQ) and a multi-laboratory intermediate precision study extending the two-laboratory robustness data presented here. Both are defined activities within the ongoing validation workplan.
Within these boundaries, the method provides a defined, validated readout that is intended to complement, not to replace, mass-based dsRNA detection within an mRNA drug substance control strategy.
Conclusion
A RIG-I–based functional assay for dsRNA detection has been developed and validated against ICH Q2(R2) acceptance criteria for specificity, range, accuracy, repeatability, intermediate precision, and robustness, across more than eight mRNA constructs and four nucleotide chemistries. The assay quantifies dsRNA species capable of engaging the RIG-I pattern recognition receptor and provides an orthogonal, biology-informed complement to existing mass-based detection methods within an mRNA drug substance control strategy.
Selected References
- Baiersdörfer, M., Boros, G., Muramatsu, H., Mahiny, A., Vlatkovic, I., Sahin, U., Karikó, K. (2019). A facile method for the removal of dsRNA contaminant from in vitro-transcribed mRNA. Molecular Therapy – Nucleic Acids, 15, 26–35.
- Hornung, V., Ellegast, J., Kim, S., Brzózka, K., Jung, A., Kato, H., Poeck, H., Akira, S., Conzelmann, K.-K., Schlee, M., Endres, S., Hartmann, G. (2006). 5′-Triphosphate RNA is the ligand for RIG-I. Science, 314, 994–997.
- ICH (2023). ICH Harmonised Guideline Q2(R2): Validation of Analytical Procedures. International Council for Harmonisation, Step 5.
- Karikó, K., Muramatsu, H., Welsh, F.A., Ludwig, J., Kato, H., Akira, S., Weissman, D. (2008). Incorporation of pseudouridine into mRNA yields superior nonimmunogenic vector with increased translational capacity and biological stability. Molecular Therapy, 16, 1833–1840.
- Karikó, K., Muramatsu, H., Ludwig, J., Weissman, D. (2011). Generating the optimal mRNA for therapy: HPLC purification eliminates immune activation and improves translation of nucleoside-modified, protein-encoding mRNA. Nucleic Acids Research, 39, e142.
- Kowalinski, E., Lunardi, T., McCarthy, A.A., Louber, J., Brunel, J., Grigorov, B., Gerlier, D., Cusack, S. (2011). Structural basis for the activation of innate immune pattern-recognition receptor RIG-I by viral RNA. Cell, 147, 423–435.
- Pichlmair, A., Schulz, O., Tan, C.P., Näslund, T.I., Liljeström, P., Weber, F., Reis e Sousa, C. (2006). RIG-I-mediated antiviral responses to single-stranded RNA bearing 5′-phosphates. Science, 314, 997–1001.
- Schlee, M., Roth, A., Hornung, V., Hagmann, C.A., Wimmenauer, V., Barchet, W., Coch, C., Janke, M., Mihailovic, A., Wardle, G., Juranek, S., Kato, H., Kawai, T., Poeck, H., Fitzgerald, K.A., Takeuchi, O., Akira, S., Tuschl, T., Latz, E., Ludwig, J., Hartmann, G. (2009). Recognition of 5′ triphosphate by RIG-I helicase requires short blunt double-stranded RNA as contained in panhandle of negative-strand virus. Immunity, 31, 25–34.
- Schönborn, J., Oberstrass, J., Breyel, E., Tittgen, J., Schumacher, J., Lukacs, N. (1991). Monoclonal antibodies to double-stranded RNA as probes of RNA structure in crude nucleic acid extracts. Nucleic Acids Research, 19, 2993–3000.
- Triana-Alonso, F.J., Dabrowski, M., Wadzack, J., Nierhaus, K.H. (1995). Self-coded 3′-extension of run-off transcripts produces aberrant products during in vitro transcription with T7 RNA polymerase. Journal of Biological Chemistry, 270, 6298–6307.
About The Author:
 Mohamad Toutounji, Ph.D., is founder and principal scientist of Molgenium, a pharmaceutical sciences consultancy based in Düsseldorf, Germany. His work focuses on CMC, analytical development, and regulatory strategy for advanced therapy medicinal products and mRNA therapeutics, across both EMA and FDA frameworks.
Mohamad Toutounji, Ph.D., is founder and principal scientist of Molgenium, a pharmaceutical sciences consultancy based in Düsseldorf, Germany. His work focuses on CMC, analytical development, and regulatory strategy for advanced therapy medicinal products and mRNA therapeutics, across both EMA and FDA frameworks.