
Rethinking The "3:1 Rule" In LNP Production
By Cedric Devos, Aniket Udepurkar, and Peter Sagmeister, Postdoctoral Associates, MIT

On the surface, lipid nanoparticle (LNPs) production can appear deceptively simple: rapidly mix the carefully selected lipids dissolved in ethanol with the therapeutic xRNA in an acidic buffer and the particles “self-assemble.” But anyone working in the field knows this step is a major bottleneck. The relationship between critical process parameters and LNP-critical quality attributes is still poorly mapped. Compounding this, translating insights from early-stage studies to manufacturing-scale remains complex.
Early-stage work typically uses microfluidic mixers to conserve material, while manufacturing more often relies on jet-based systems, most commonly confined impinging-jet mixers that collide two streams at high speed. Despite these differences in equipment, nearly everyone in RNA research circles follows the same recipe: a 3:1 flowrate of aqueous (cargo) to organic (lipid) phases. It’s a rule of thumb that has guided the field for years, and practically no one has questioned.
A team in the Chemical Engineering Department at MIT recently asked a simple question: if LNPs form through rapid mixing, why is the industry using the same 3:1 ratio instead of operating under more ideal mixing conditions found with a 1:1 flow-rate ratio? Over the course of their research, they confirmed the industry’s default 3:1 flow-rate ratio (aqueous:organic) exists for a reason: operating at 1:1 produces LNPs that rapidly evolve and show poor RNA encapsulation, making 1:1 unsuitable for practical manufacturing.
A systematic, parametric study showed that at 3:1, the process is robust: most input parameters have little effect on the final particle attributes. This sounds promising in principle, but it also means there is little tuneability for parameters that directly affect therapeutic performance, such as particle size and internal morphology. As a result, the process leaves limited room to shape attributes that matter most. Only the total flow rate, which directly sets the mixing intensity, meaningfully shifts LNP properties, and even that lever is constrained. Slowing the mixing to gain tunability comes at a steep cost – encapsulation efficiency collapses and the manufacturing costs increase sharply.
At 1:1, however, relationships react differently and expose the underlying physics of LNP self-assembly. Despite the instability, other parameters such as buffer strength, pH, and N/P ratio exerted far stronger influence on size, structure, and encapsulation. In other words, the region where LNPs are unstable is also the region where the process is most sensitive, providing a window into how formulation and process variables shape particle self-assembly.
These findings raised another fundamental question: if the most sensitive, tunable region sits on the edge of instability, could the Team, and others, briefly operate there, extract the benefits, then rapidly stabilize the particles before they degrade?
Designing LNPs by Capturing Them at the Right Moment
The Team demonstrated that answer is, “Yes.”
By initiating assembly at a 1:1 ratio, where size and morphology are highly responsive to buffer strength, pH, N/P ratio, and other inputs, then swiftly driving the system back to the conventional 3:1 conditions, particles can be stabilized at precisely the desired state. The key is to arrest the LNPs’ evolution at precisely the right moment, enabling the precise engineering of LNP size and morphology.
In practice, this was achieved by connecting two impinging-jet mixers in a series, connected by tubing of a defined volume that set the residence time in the millisecond range. Crucially, both mixers were run under ideal mixing conditions (at 1:1 and 2:2 ratio), ensuring rapid quenching and consistently high encapsulation efficiency. Think of the first jet-mixer as the start of the self-assembly, followed by a phase in which LNPs evolve in different sizes and shapes, and the second jet-mixer as the “freeze” button.
More importantly, once both jet mixers were operated under ideal mixing conditions, the system became predictable. The evolution of size and morphology could be modeled, allowing the team to select exactly when, and under which conditions, the second jet mixer should quench the process. For example, increasing the residence time (effectively shifting the “when” in the assembly trajectory), allowing the particles to evolve longer in the sensitive regime, led to a predictable increase in mean particle size. The shape, or more precisely the fraction of LNPs displaying non-circular, blebbed morphologies rose with longer residence times. The process offered a broad design space for simultaneously shaping size and morphology, because multiple input parameters can be tuned independently.
This two-step method is highly predictable, but also more straightforward to track, model, and control. Additionally, the approach was demonstrated across different lipid compositions and formulation conditions. Once ideal jet-mixing is ensured, the same principles hold regardless of the ionizable lipid structure or helper lipid ratios, underscoring it as a platform method, not a one-off formulation “trick.”

Figure 1 - Demonstration of the different LNP shapes that can be obtained with the novel two-step method.
Deliberately Engineered LNPs: Why They Matter
Gaining real control over LNP assembly means shape and size can be selected to match the biological target, without sacrificing RNA through poor encapsulation (shown in Fig. 1). This directly improves potency and reduces costs. More predictable particle properties also simplify downstream purification and quality control, which also remain major cost and time bottlenecks in mRNA manufacturing.
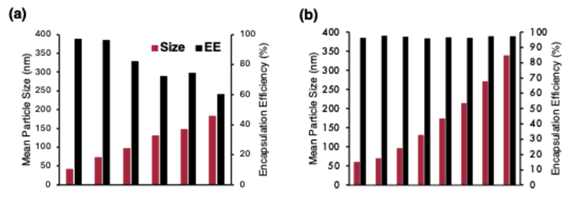
Figure 2 - Comparison of mean particle size and corresponding encapsulation efficiency between the conventional manufacturing approach and the new two-stage, ideal-mixing process.
Taken together, the Team’s work marked a transition from empirical “mix and hope” practices toward deliberately engineered LNPs, where morphology, size, and encapsulation are set by defined process conditions rather than by trial-and-error. Instead of relying solely on lipid redesign or new chemistries, the study showed that precise control over the physics of self-assembly provided a powerful new lever to shape the desired particles. Their research opens the door to next-generation particle designs tailored to specific tissues, administration routes, and intracellular delivery pathways, a foundation for more effective and more affordable RNA medicines.
The Team’s full findings were published on September 15, 2025, in ACS Nano and supervised by Richard D. Braatz, PhD, and Allan S. Myerson, PhD.
About The Experts
 Cedric Devos is a postdoctoral associate in the Department of Chemical Engineering at the Massachusetts Institute of Technology (MIT), where he has worked since 2023 under Professor Allan S. Myerson on lipid nanoparticle delivery for nucleic acid therapeutics. He received his BSc (2017) and MSc (2019) in chemical engineering from KU Leuven, where he later completed his PhD (2023) as an FWO Fellow under Professor Simon Kuhn. His doctoral work was complemented by a visiting research stay at Janssen Pharmaceuticals (Johnson & Johnson). His research spans crystallization, nanoparticle formulation, and process intensification.
Cedric Devos is a postdoctoral associate in the Department of Chemical Engineering at the Massachusetts Institute of Technology (MIT), where he has worked since 2023 under Professor Allan S. Myerson on lipid nanoparticle delivery for nucleic acid therapeutics. He received his BSc (2017) and MSc (2019) in chemical engineering from KU Leuven, where he later completed his PhD (2023) as an FWO Fellow under Professor Simon Kuhn. His doctoral work was complemented by a visiting research stay at Janssen Pharmaceuticals (Johnson & Johnson). His research spans crystallization, nanoparticle formulation, and process intensification.
 Aniket Udepurkar is a postdoctoral associate in the Department of Chemical Engineering at the Massachusetts Institute of Technology (MIT), working with Professor Allan S. Myerson on mRNA and lipid nanoparticle formulation in a program supported by the FDA. He earned his B.Tech. in chemical engineering from the Indian Institute of Technology Guwahati (2015) and his MSc (2019) and PhD (2024) in chemical engineering from KU Leuven under Professor Simon Kuhn. His doctoral work focused on ultrasonic microreactors for the synthesis of functional nanoparticles, complemented by a research visit to Professor Klavs F. Jensen’s group at MIT. His interests include nanoparticle engineering, flow chemistry, and the development of intensified, scalable processes for therapeutic delivery systems.
Aniket Udepurkar is a postdoctoral associate in the Department of Chemical Engineering at the Massachusetts Institute of Technology (MIT), working with Professor Allan S. Myerson on mRNA and lipid nanoparticle formulation in a program supported by the FDA. He earned his B.Tech. in chemical engineering from the Indian Institute of Technology Guwahati (2015) and his MSc (2019) and PhD (2024) in chemical engineering from KU Leuven under Professor Simon Kuhn. His doctoral work focused on ultrasonic microreactors for the synthesis of functional nanoparticles, complemented by a research visit to Professor Klavs F. Jensen’s group at MIT. His interests include nanoparticle engineering, flow chemistry, and the development of intensified, scalable processes for therapeutic delivery systems.
 Peter Sagmeister is a postdoctoral associate in the Department of Chemical Engineering at the Massachusetts Institute of Technology (MIT), where he works on advancing pharmaceutical manufacturing through data-driven process development and scalable technologies. He received his BSc (2012) and MSc (2018) from the University of Graz and completed his PhD (2022) under Professor Oliver Kappe, specializing in continuous flow chemistry and digital tools for active pharmaceutical ingredient production. Before joining MIT, he conducted postdoctoral research at the University of Graz and the Research Center of Pharmaceutical Engineering, contributing to publications and patents on process analytical technology and machine learning for small-molecule synthesis. His work focuses on integration of automation, modeling, and flow processing for next-generation drug manufacturing.
Peter Sagmeister is a postdoctoral associate in the Department of Chemical Engineering at the Massachusetts Institute of Technology (MIT), where he works on advancing pharmaceutical manufacturing through data-driven process development and scalable technologies. He received his BSc (2012) and MSc (2018) from the University of Graz and completed his PhD (2022) under Professor Oliver Kappe, specializing in continuous flow chemistry and digital tools for active pharmaceutical ingredient production. Before joining MIT, he conducted postdoctoral research at the University of Graz and the Research Center of Pharmaceutical Engineering, contributing to publications and patents on process analytical technology and machine learning for small-molecule synthesis. His work focuses on integration of automation, modeling, and flow processing for next-generation drug manufacturing.