
7 New MHRA Guidances To Help You With Decentralized Manufacturing For Cell And Gene Therapies
By Peter H. Calcott, Ph.D., FRSC, president and CEO, Calcott Consulting LLC

In 2025, the MHRA issued a change to the regulations for licensure of medicines employing cell and gene therapy technology.1 This change is particularly relevant to those treatments that require a significant manufacturing activity outside the traditional manufacturing plant, especially at the bedside of patients. The building blocks of these autologous pharmaceuticals are manufactured in classical plants but their effectiveness requires harvesting a patient’s cells or tissues, the treatment of those cells or tissues with these building blocks very close to the patient, and administration of the resultant product to that patient. These activities outside the classical production plant were always considered too complex to be performed without the drug owner in attendance. To understand the difficulty of this type of technology, the reader is directed to a review in 2024 by the author.2 Further, the implications of these regulatory changes were discussed in an article published in early 2025.3
Reviewing these regulatory changes is quite an exhausting process, requiring going through the regulations line by line, so the MHRA promised that the changes to these regulations would be followed by guidances to aid industry.4 This article will focus on the seven guidances that were published in June 2025:
- Decentralized Manufacturing – the Designation Step
- Decentralized Manufacturing – Marketing Authorization application
- Decentralized Manufacturing – Clinical Trial Authorization (CTA) and Good Clinical Practices
- Decentralized Manufacturing – U.K. Guideline of Good Pharmacovigilance Practices
- Decentralized Manufacturing – Guidance on Good Manufacturing Practices (GMP)
- Decentralized Manufacturing - Labelling
- Decentralized Manufacturing – Modular Manufacturing and Point of Care regulations 2025: Overview
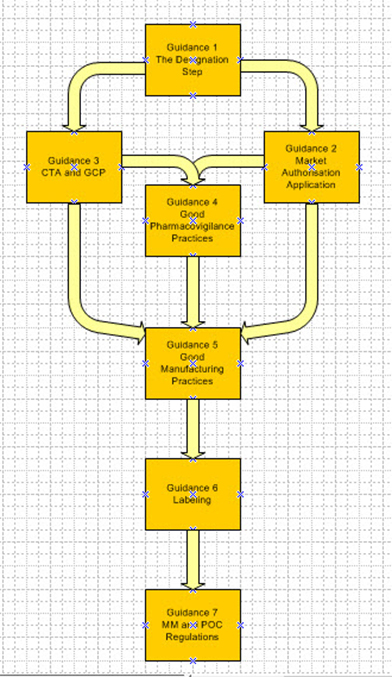
Figure 1: Interrelationship between the guidances
1. Decentralized Manufacturing – The Designation Step
This guidance describes the process whereby the sponsor petitions the MHRA for evaluation of the product proposed to use the decentralized manufacturing process. This evaluation is to determine whether the product can use this process and whether it meets the requirements for the point of care (POC) or modular manufacturing (MM) concept. This should happen early in the development cycle. The applicant should present data on the program to justify the specific approach, including quality and clinical data, as appropriate. This can be for a new application (Clinical Trial Authorisation [CTA] or Marketing Authorisation [MA]) or via an amendment to a preexisting CTA or variation to a MA. This process also can apply to “specials” (one-off medicines).
What constitutes POC or MM designations?
POC products are designated as those medicinal products that, for reasons relating to method of manufacture, shelf life, constituents, or method or route of administration, can only be manufactured at or near the place where the product is to be used or administered. The guidance further states that convenience and cost should not be criteria.
MM are defined as medicinal products that, for reasons relating to deployment, the licensing authority determines it necessary or expedient to be manufactured or assembled in a modular unit. For this application, the applicant should present evidence justifying this route related to a public health requirement and/or significant clinical advantage, improving efficacy, tolerability, or safety.
The preliminary decision could be as soon as 30 days, with full approval in 60 days, assuming all information is presented.
2. Decentralized Manufacturing – Marketing Authorization Application (MAA)
With the positive opinion from the designation step in hand, the MAA can proceed with adequate reference to the designation. If the designation is not in hand but a favorable outcome is expected, the MAA also can proceed with caution. Failure to close out the designation before review of the MAA can result in rejection.
The expectations for these MAAs are similar to those for conventional products. Particular emphasis is placed on process validation and demonstration that there is comparability between products made at the variety of the remote manufacturing sites. Since a lot of autologous products use real time release testing (RTRT), particular emphasis should be placed on the data to support this strategy.5 These MAAs describe the controlling site where the majority of the manufacture occurs. It also describes how it will oversee the decentralized sites. The submission also includes a Decentralized Manufacturing Master File (DMMF), which describes how to complete the manufacturing at the decentralized site.
3. Decentralized Manufacturing – Clinical Trial Authorization (CTA) and Good Clinical Practices (GCP)
A CTA should also, like the MAA, have adequate reference to the DM designation. A reference to the application for designation should be included and should indicate that the designation has not been changed. A “control site,” which may be the site where the components are made, must be designated. A Manufacturing Importation Authorisation Application (MIA) should be submitted with supporting data.
The requirements of product development should follow standard methods used for conventional medicinal products. Emphasis on RTRT should be presented as described for MAA applications. Because blinded trials are common in development, emphasis should be made on the mechanism of blinding and retaining the blind during the trial.
For POC trials, special emphasis should be paid to assuring retention of blinding in such areas as:
- process required to manufacture dosage form
- time taken to manufacture or prepare the dose
- difference in appearance or other characteristics, such as temperature or equipment required, between active and placebo products
- process required for administration of dose
- record keeping, especially those associated with manufacture and release of investigational medicinal product (IMP).
Like the MAA described in guidance 2, there will be a defined control site with operations and oversight of the remote sites. A DMMF also will be included with instructions on how to finish the manufacturing steps.
4. Decentralized Manufacturing – U.K. Guideline of Good Pharmacovigilance Practices
MHRA recognizes that since some manufacture occurs outside a conventional manufacturing plant, the coordination of safety and efficacy evaluation impacted by the manufacture is a significant challenge. With a significant number of sites of manufacture that could interpret manufacturing instructions differently, the pharmacovigilance program must be acutely sensitive to these subtle differences and be able to track them effectively. To assure this, product traceability becomes a critical element in this program. This traceability is not just important for safety monitoring but to assure the right medicine gets to the right patient.
The general principles of pharmacovigilance for conventional products apply to DM.6 Heavy emphasis on a risk management plan is paramount. Where additional risk management measures are warranted, these should be addressed. Traditional areas such as signal management and periodic safety updates should be managed commensurately with the risk involved for these unique products. Some of these products may require additional monitoring. Because these approaches are new, special emphasis should be made to assure product safety information is updated expeditiously.
There should be a robust quality management system with a designated QP for pharmacovigilance to oversee the program. A Pharmacovigilance System Master File should be prepared and kept up to date. Early Access to Medicines Scheme (EAMS) brings medicines to patients quickly but the risk should be considered to assure adequate monitoring of patients. The same concerns are shared with “specials.”
The take-home message is that all pharmacovigilance principles apply and are expected but because of the novelty of these products and approach, caution should be exercised and anticipated.
5. Decentralized Manufacturing – Guidance on Good Manufacturing Practices (GMP)
The control site must be located in the U.K. and have a manufacturing license. At that site there should be a functional QMS and have procedures to support the following activities:
- management of DM control strategy
- generation and management of DMMFs used to assure other sites operate adequately, following classic DMF rules of designation and reporting
- onboarding, suspending and pausing remote sites with adequate communication methods to these sites
- ongoing oversight of these sites including data integrity
- assurance of calibration and maintenance of equipment
- supplying materials and components to the sites
- a training program for the sites
- product release procedures for POC products
- a QP for oversight
- a completed DMMF in place.
The program should assure that the remote sites are functioning adequately. During inspections, these assurances will be a prime focus. An authority might inspect a selection of remote sites but not necessarily all. These sites and their approval are kept at the control site for inspection and are not submitted to the agency.
The QMS should be comprehensive and treat the remote sites like a CMO with oversight, audits, and quality agreements. The guidance gives a comprehensive set of requirements covering all aspects of quality.
6. Decentralized Manufacturing – Labelling
All requirements defined for conventional medicines apply and must be met. There is only one exception: POC medicines that are required to be administered immediately, with none retained for subsequent administration. These do not have to be labeled after manufacture. However, MHRA encourages pre-labeling containers. What constitutes “immediate”? Two minutes!
7. Decentralized Manufacturing – Modular Manufacturing and Point of Care Regulations 2025: Overview
By far the longest guidance, number 7 is a full 47 pages and comprehensively covers several topics including legislation and regulations with definitions. It references the human medicines regulations (2012) in section 2 and clinical trials regulations (2004) in section 3, together with a glossary of legislation taking you line by line with the changes.
Section 2 covers 14 subtopics related to commercial medicinal products, including interpretation (with definitions of acronyms), manufacture, exemptions, applying for a license, obligations in manufacture, qualified person, variations to a license, variations of a POC or MM master file, applications for MAs, materials to accompany applications, pharmacovigilance, specials requirements, EAMS, and labeling.
Section 3 mirrors section 2 but is focused on IMP used in clinical trials. The 12 subtopics include the following: interpretation (with definitions of acronyms), supply of IMP for clinical trials, requirements to manufacture or import, exemptions, applying for a manufacturing authorization, considerations for application of manufacturing authorization, standard provisions for authorizations, variation of manufacturing authorization, variation of a POC and MM master file, labeling, infringement notices, and offenses.
Both these sections illustrate and clarify the various requirements that must be met. Practitioners of this innovative technology should read the guidances in depth and assure they are meeting the substance as well as the intent of these regulatory documents. Reading through them, it is clear that this bold move from the MHRA to “relax” control from regulators to the license holder is an experiment in progress. They have tried to think through as many situations and technical challenges and put in place controls to assure a smooth rollout of the program. Have they identified them all? Perhaps not, but based on a reading of these guidances, they have identified a large number. This bold move has been attempted while they are still working on Brexit and separating their regulations from the EMA. Hats off to the MHRA.
References
- Statutory Instrument laid in Parliament provides first regulatory framework of its kind that will transform the manufacture of innovative medicines at the point of patient care (2025) https://www.gov.uk/government/news/statutory-instrument-laid-in-parliament-provides-first-regulatory-framework-of-its-kind-that-will-transform-the-manufacture-of-innovative-medicines-at
- Strategies to Tackle CAR-T Product Challenges. 2024 https://www.cellandgene.com/doc/strategies-to-tackle-car-t-product-challenges-0001
- MHRA Issues New Regulation on Modular and Point of Care Manufacture of ATMPs (2025)
- Introduction to the UK’s Guidelines on Decentralised Manufacturing – Decentralised Manufacturing Hub (2025) https://www.gov.uk/government/collections/decentralised-manufacture-hub?utm_medium=email&utm_campaign=govuk-notifications-topic&utm_source=4fa84da5-90b5-472e-8ec3-16b7253df773&utm_content=immediately#guidance
- Guideline on Real Time Release Testing (formerly parametric release) https://ema.europa.eu/en/rel-time-release-testing-scientific-guideline and EudraLex Vol 4 Annex 17 on ‘RTRT and Parametric Release https://health.ec.europa.eu/document/download/78d31fc9-760f-4fe9-9ccd-95595ef48a71-en?filename=2018-annex17-en.pdf
- Good Pharmacovigilance practices (GVP) – European Medicines Agency (EMA) https://www.ema.europa.eu/en/human-regulatory-overview/post-authorisation/pharmacovigilance-post-authorisation/good-pharmacovigilance-practices-gvp
About The Author:
 Peter H. Calcott, D.Phil., is president and CEO of Calcott Consulting LLC, which delivers solutions to pharmaceutical and biotechnology companies in the areas of corporate strategy, supply chain, quality, clinical development, regulatory affairs, corporate compliance, and enterprise e-solutions. He has also served as an expert witness. He also teaches at the University of California, Berkeley in the biotechnology and pharmaceutics postgraduate programs. Previously, he was executive VP at PDL BioPharma, chief quality officer at Chiron and Immunex Corporations, and director of quality assurance for SmithKline Beecham and for Bayer. He has also held positions in R&D, regulatory affairs, process development, and manufacturing at other major pharmaceutical companies. He has successfully licensed products in the biologics, drugs, and device sectors on all six continents. Calcott holds a doctorate in microbial physiology and biochemistry from the University of Sussex in England. He has been a consultant for more than 20 years to government, industry, and academia.
Peter H. Calcott, D.Phil., is president and CEO of Calcott Consulting LLC, which delivers solutions to pharmaceutical and biotechnology companies in the areas of corporate strategy, supply chain, quality, clinical development, regulatory affairs, corporate compliance, and enterprise e-solutions. He has also served as an expert witness. He also teaches at the University of California, Berkeley in the biotechnology and pharmaceutics postgraduate programs. Previously, he was executive VP at PDL BioPharma, chief quality officer at Chiron and Immunex Corporations, and director of quality assurance for SmithKline Beecham and for Bayer. He has also held positions in R&D, regulatory affairs, process development, and manufacturing at other major pharmaceutical companies. He has successfully licensed products in the biologics, drugs, and device sectors on all six continents. Calcott holds a doctorate in microbial physiology and biochemistry from the University of Sussex in England. He has been a consultant for more than 20 years to government, industry, and academia.